
Article co-rédigé avec Thierry Candresse (INRAE)
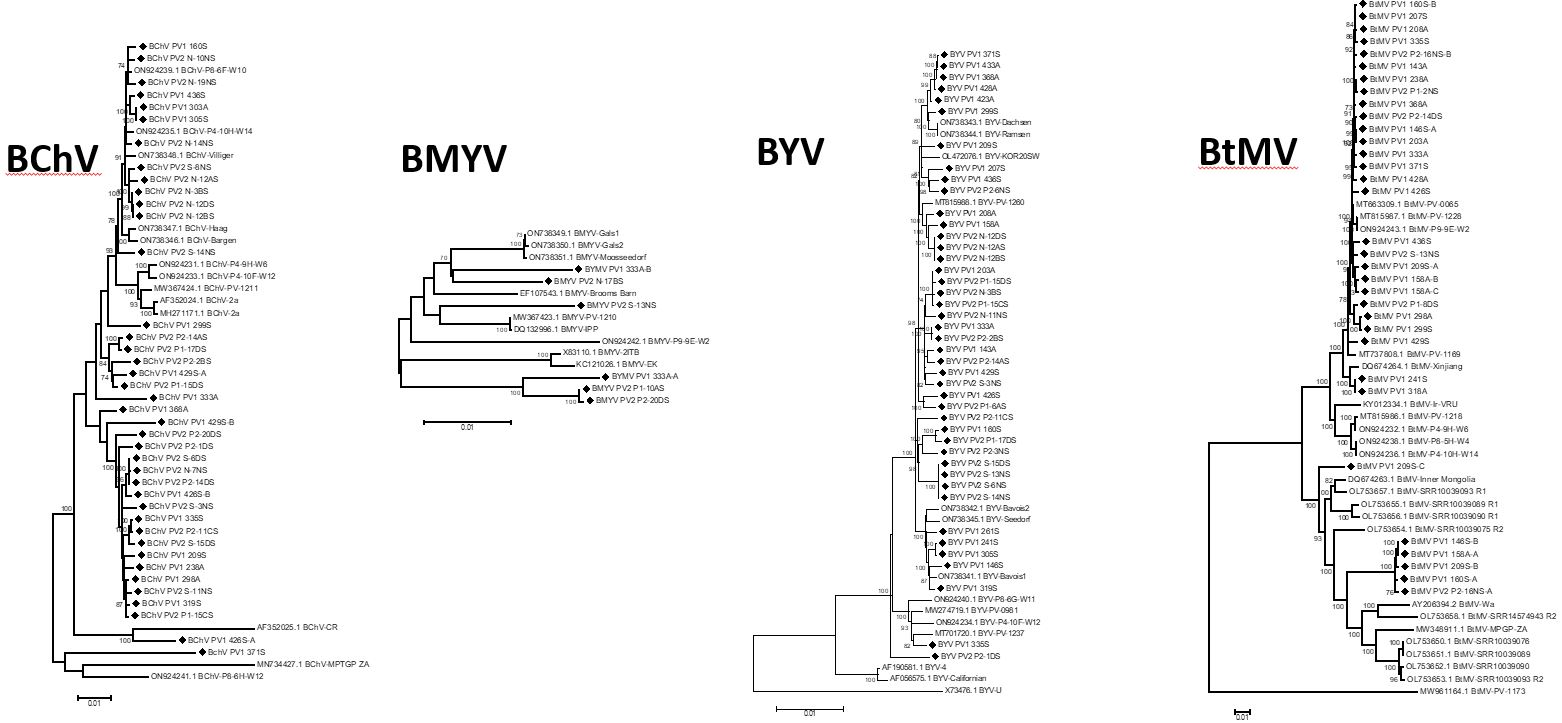
Les séquences génomiques complètes des isolats viraux présents dans les échantillons prélevés en France ont été analysées dans le cadre du projet Provibe du PNRI puis l’analyse a été élargie avec des séquences d’isolats disponibles dans les bases de données internationales. La diversité des séquences génomiques peut être analysée à l’aide d’arbres phyllogénétiques comme illustré ci-dessus : la distance génétique entre isolats viraux est proportionnelle à la longueur des branches de l'arbre. Sur ces arbres, les isolats des betteraves prélevés en France sont indiqués avec un figuré noir, ceux sans figuré sont issus des bases internationales. Le tableau suivant synthétise les résultats de diversité génétique (distance génétique moyenne entre paires d'isolats pris au hasard).
Diversité génétique des différents virus, calculée pour la France seule ou pour l'ensemble des isolats viraux pour lesquels des séquences génomiques complètes sont disponibles :
| Echantillons français | Echantillons français + bases internationales | |
| BMYV | 3,1% | 3,1% |
| BChV | 2,3% | 2,8% |
| BYV | 0,6% | 1% |
| BtMV | 3,4% | 5,8% |
Si l'on compare à d'autres genres viraux, ces données montrent qu'il n'existe qu'une faible diversité génétique parmi les isolats français pour les 4 virus responsables des jaunisses de la betterave. Même à l’échelle internationale, la diversité reste faible voire très faible. Cette relative homogénéité des virus, sans diversification géographique, suggère qu’ils circulent sur de grandes distances grâce aux pucerons vecteurs. Par ailleurs, les données du projet Provibe montrent qu'il est fréquent d'observer au sein d'une même parcelle des isolats significativement différents, suggérant l'existence de multiples sources de contamination, ou de réservoirs hébergeant simultanément différents isolats viraux.






